

ML Achieves Promising Results in Drug Discovery, Even With Limited Data
Traditionally, designing new drugs is inherently complex. The drug discovery process starts by identifying what causes a disease, then making a list of drug candidates, and finally subjecting them to virtual screening. Thanks to recent advances in machine learning (ML) and more available data, we're now better equipped to solve complex problems in a data-driven manner.
Screening or generative strategies
In the ML field, research groups typically approach molecular design challenges in two ways:
- Screening: Review a list of molecules and check their properties. ML models are trained to establish structure-to-property relationships.
- Generative: Create new molecule designs iteratively, using feedback from predictive models (from the screening scenario) to judge the quality of the candidates.
Both methods explore target properties and find potential candidates. However, the lack of data on certain properties can affect the robustness of predictive models.
This article shares the impact of advanced ML methods and partnerships on drug discovery. ML techniques can help identify novel compounds, especially when there's not much data available. Our hypothesis is that compounds with similar structures tend to have similar functions. We've put this into practice by identifying novel coagulant candidates that help control blood clotting.
Improve blood clot treatment with autoencoders
To achieve this, we use autoencoders. These help us assign each molecule its own coordinates in a ‘chemical space’ and analyze known coagulants and anticoagulants that are distributed in that space.
Besides the technical challenge, blood clotting control is important for treating patients after surgery and those dealing with blood clotting disorders (anticoagulation/coagulation cascades) like thrombosis or hemophilia.
Treatment varies by case. You can add missing proteins (substitution therapy) or correct the (anti)coagulation balance. This could mean inhibiting certain proteins or blocking their production. For example, suppressing anticoagulant proteins enhances coagulation activity. The reverse works too.
Autoencoders: A type of AI that learns to compress and then reconstruct data, often used to find patterns or reduce noise in information.
Protein C: A protein in your blood that prevents excessive clotting, ensuring your blood flows smoothly.
Thrombin: An enzyme that helps your blood clot when you get a cut or an injury, stopping the bleeding.
Create and analyze datasets
For autoencoders to work best, you need a strong dataset. To build the inhibition dataset, we used the publicly accessible BindingDB database (accessed June 2023). This database gives protein and ligand structures in Simplified Molecular Input Line Entry System (SMILES) format. It also shows how strongly the protein and ligand interact. We pulled two types of data from BindingDB:
- Activity class (active, inactive)
- Activity measure (inhibition constant Ki)
We focused on thrombin and protein C, which both control blood clotting. These two belong to coagulation and anticoagulation cascades, respectively. The thrombin classification dataset had 5,009 ligands, where 3,323 actives based on Ki, Kd, EC50, or IC50. The thrombin regression dataset had 2,270 ligands with available Ki values, and 1,927 of these were active (having Ki ≤ 10,000 nM).
The protein C classification dataset had 188 protein C ligands with 103 actives. Ki is available for 125 ligands, and 79 are confirmed active protein C inhibitors. The number of known protein C inhibitors was too low to train confident and robust ML models. But there was good news as molecules with known activities are not needed to train the autoencoder. The autoencoder's goal is to learn the universal chemical rules of SMILES syntax. This training used large sets of valid SMILES examples.
Generate new molecules
With a detailed dataset in hand, we started to create new molecules. Our approach has two key stages:
- First, we built an autoencoder to map the chemical space of small organic molecules.
- Then we generated candidates by exploring this mapped space.
Autoencoders are an unsupervised method trained to match outputs with inputs. They first compress input information into a bottleneck and then decompress it back to output. Each data sample comes with its own embedding vector, acting as coordinates in a hyperspace. This applies to molecules too – each can be encoded with unique coordinates (embedding vector) in a chemical hyperspace.
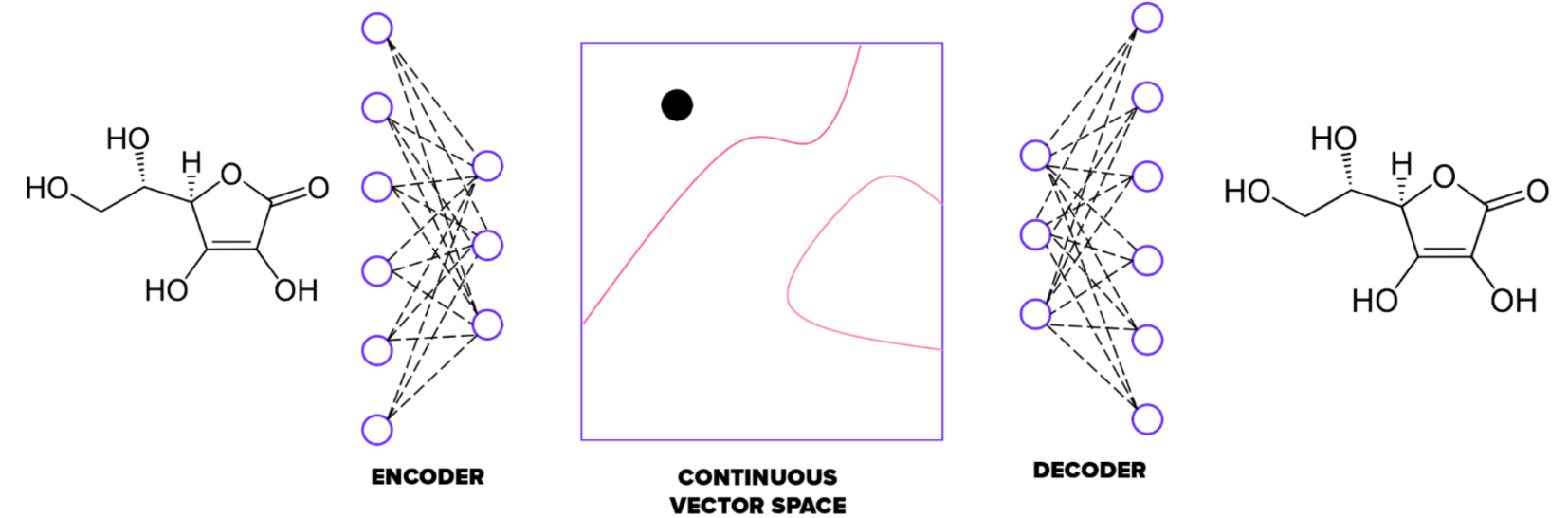
The autoencoder has two parts: an encoder and a decoder. The embedding vector is derived from the encoder's output. In our implementation, the size of the embedding is 100. We fine-tuned the autoencoder by changing the number of neural layers, their types, shapes, and activation functions. As a result, the encoder has four convolutional and one fully connected layer. The decoder mirrors this structure with one fully connected and four convolutional layers.
- Thrombin affinity predictors: As data availability for known thrombin inhibitors is high, we trained corresponding predictors and validated the generation of anticoagulants. We used a two-stage assessment approach, predicting an inhibitory class first (active vs. nonactive) and then, if active, the inhibition constant (Ki), using ensembles of different ML regression models.
- Cluster thrombin and protein C inhibitors: In the autoencoder's embedding space, cluster analysis identified several clusters for protein C and thrombin inhibitors. By comparing the characteristic distances inside and between these clusters, we chose a "safe" radius of (dsep) = 0.8. Like the coordinates in the embedding space, this distance is dimensionless. The radius is intended to cover known inhibitors while avoiding the inclusion of inhibitors from the opposite protein.
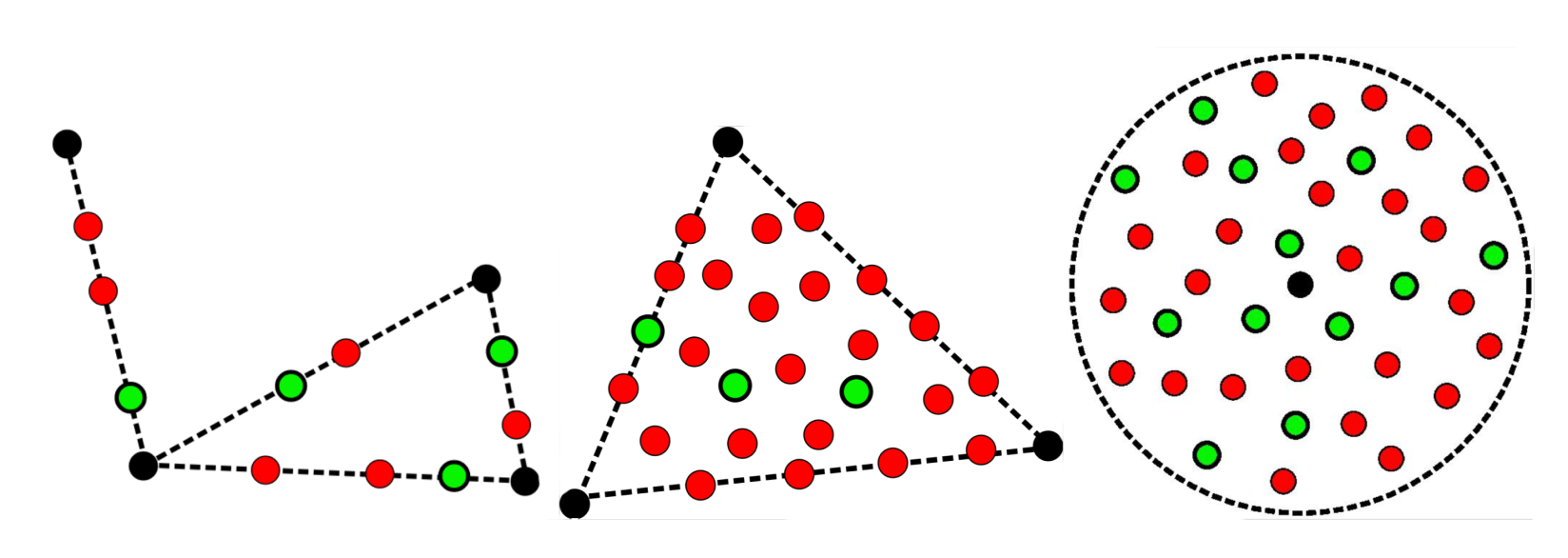
- Generate new molecule candidates: We tried a few strategies to generate new compounds: We interpolated between pairs of reference compounds or picked points within triangle-like domains defined by triplets of compounds. We also drew an imaginary hypersphere around a reference compound to find new candidate molecules within that space. All the generated embeddings were:
- Decoded to SMILES strings
- Tested on the chemical validity
- Filtered based on the proximity to desired or undesired inhibitor groups
Filled black circles show the known inhibitors. Red circles, which represent incorrect SMILES, are filtered out. The correct ones, in green, are kept and undergo a set of filters to rank and prioritize them.
- MegaMolBART: We included MegaMolBART, a transformer-based language model, alongside our autoencoder-based approach. This solution is based on the BART (Bidirectional and Auto-Regressive Transformer) architecture. It's a generative AI (Gen AI) model developed through a partnership between AstraZeneca and NVIDIA. MegaMolBART solves a task similar to that of our autoencoder-based approach. Its interface enables generation through interpolation or by searching near reference compounds. Essentially, our approach and MegaMolBART use the same generative techniques but have unique molecular embeddings.
Generated protein C inhibitor candidates: We used the interpolation approach to generate 200,000 embeddings and yielded 5,736 correct SMILES. After we removed the original thrombin inhibitors, 5,354 unique molecules remained. We filtered with distances to the nearest known thrombin inhibitors and nearest strong protein C inhibitors, and 3,150 unique SMILES were accepted, with 1,420 predicted as active.
We created hyperspheres with radii up to a “safe” distance of dsep = 0.8 around each of the 2,270 thrombin ligands. This method yielded 34,957 unique and chemically correct structures. Of these, 16,027 unique SMILES met our acceptance criteria based on the previously mentioned distances, with 6,369 predicted as active. When we analyzed the predicted log10 Ki values for these generated compounds to the actual log10 Ki values of reference inhibitors, we found that the predicted log10 Ki values stay well below the activity threshold if we consider the reference inhibitors with log10 Ki below two.
Generated protein C inhibitor candidates: The dataset of protein C ligands had 103 active compounds. We narrowed the list of reference inhibitors to those with a log10 Ki under two and selected 15 reference strong inhibitors. With these, we generated 1,571 protein C inhibitor candidates with the hypersphere and interpolation approaches. We also ranked the generated compounds by their Synthetic Accessibility Scores (SAS), with the lowest SAS at 3.86 from the hypersphere search.
The SAS values for generated protein C inhibitor candidates passing through the Ghose, Veber, or Egan drug-likeness filters are greater than four. Interesting to note that a particular candidate with an SAS of 6.28 is common to all generative techniques (shown below in the left).
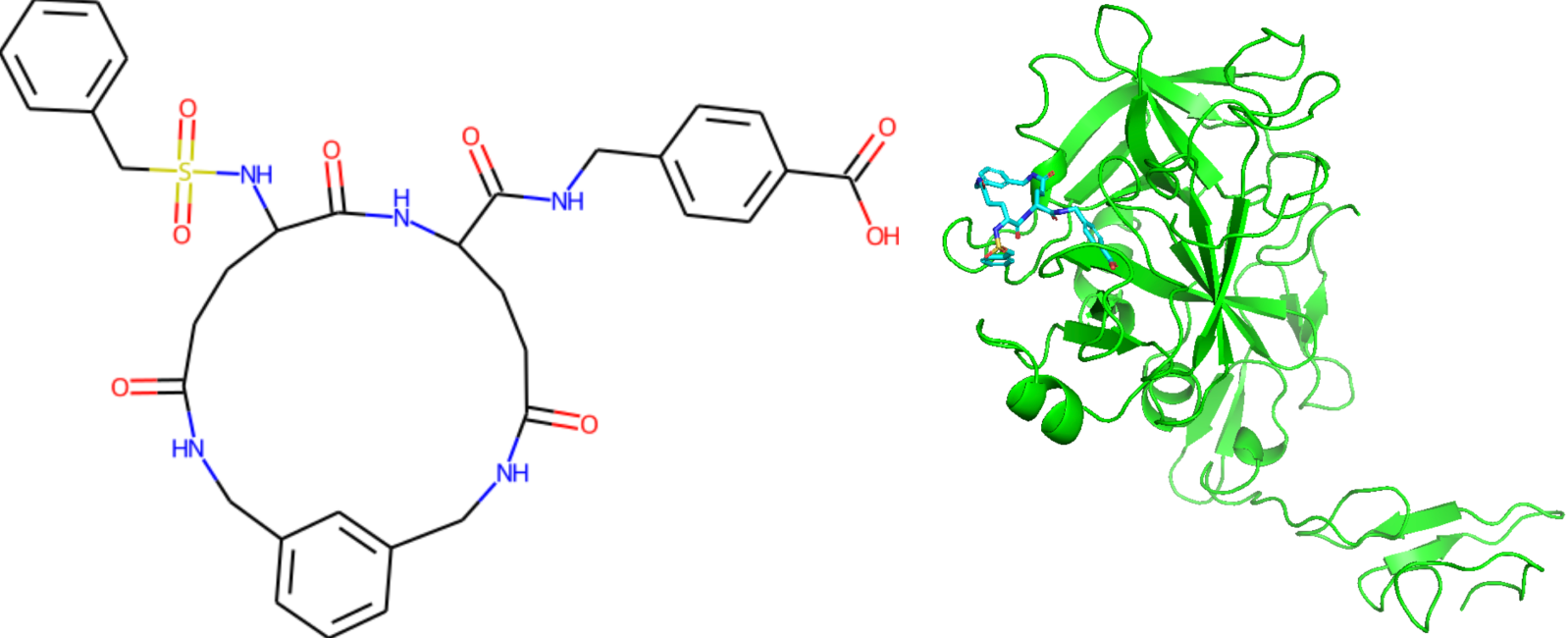
We used NVIDIA's BioNeMo a generative AI platform for drug discovery, specifically its diffusion generative model, DiffDock, to dock this molecule to the activated form of protein C. We found that the top scored pose of the candidate molecule is in the active site of the protein (shown on the right). We also used MegaMolBART to generate new molecules for protein C inhibitor pairs and around specified inhibitors. We also performed the interpolation between pairs (100 molecules per pair) and hypersphere generation around some inhibitors (up to 60 molecules per inhibitor).
A comparison: Autoencoder vs. MegaMolBART
In this work, we present our approach to suggest new inhibitor candidates by developing a deep learning model autoencoder to generate novel coagulant candidates. We compared our approach to NVIDIA’s MegaMolBART and found that it also was able to generate candidate compounds based on slightly different principles. We noticed some differences between our method and MegaMolBART. MegaMolBART tends to generate more molecules between close inhibitors than our method. For instance, it generated 14 molecules for one pair, while our method produced six. This trend is seen in other pairs as well. Our approach keeps the preserves of the reference compounds better. On the other hand, cyclopropane rings (![]() ) and cyclobutadiene rings (
) and cyclobutadiene rings (![]() ) appear in our results but are missing from the original reference inhibitors. Below is a comparison of an interpolation between two protein C inhibitors from our approach.
) appear in our results but are missing from the original reference inhibitors. Below is a comparison of an interpolation between two protein C inhibitors from our approach.
We strongly feel that the success of both generative approaches serves to provide more options for generating suggested candidates and offers promise for the future of drug discovery.
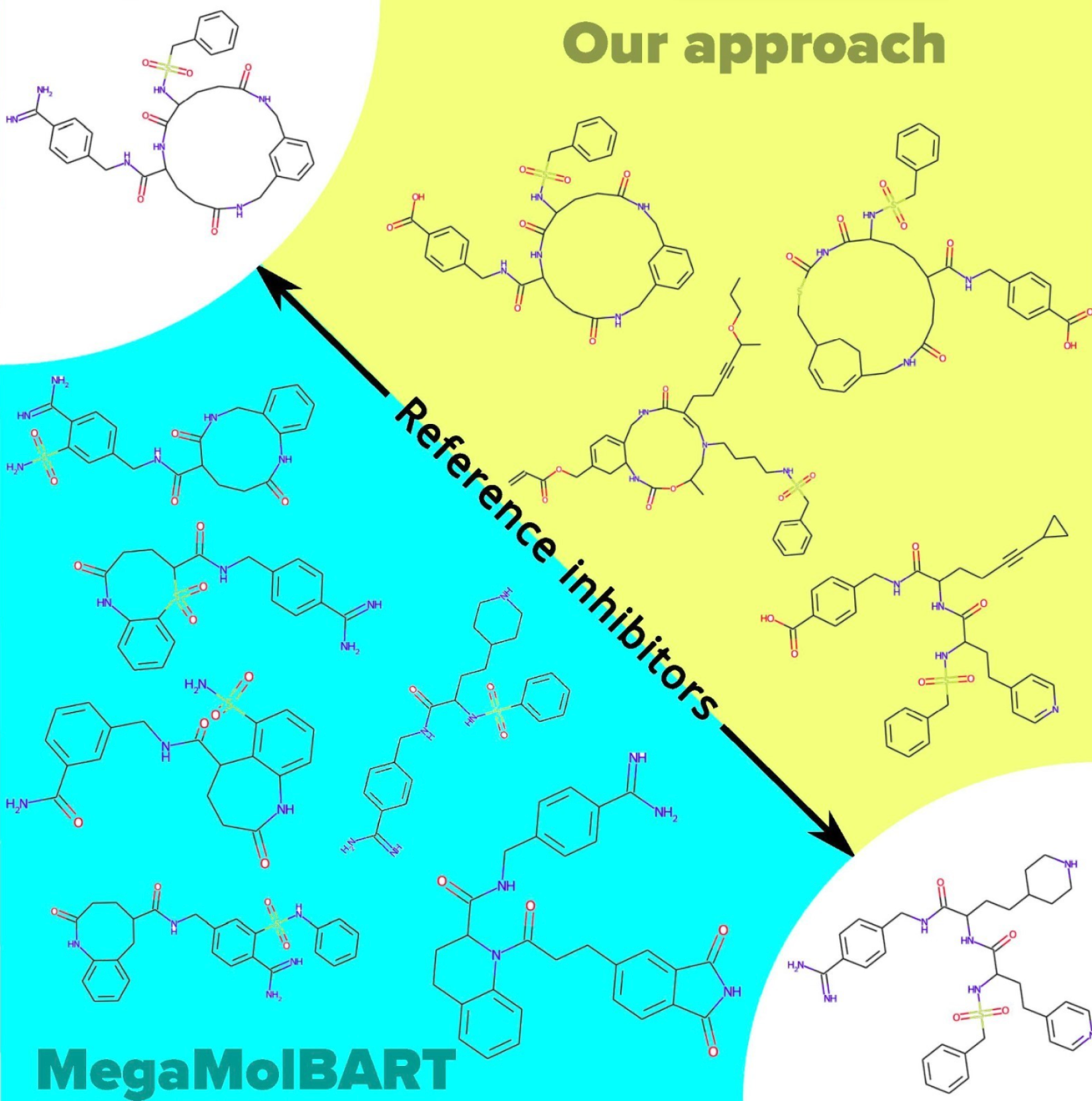
The large ring seen in the top left reference inhibitor doesn't appear in the MegaMolBART results.
Why SoftServe
At SoftServe R&D, we can help build similar pipelines for projects that may have limited data and fine-tune using our own proprietary data. Our unique expertise and elite partnership with NVIDIA provide us access to the latest tools in the field.
Read the full study to see how ML sets new standards in drug discovery and achieves promising results, even with limited data.